Markus KowalewskiUniversitetslektor
Om mig
The group works on a wide variety of topics involving photo chemistry, coherent control, polaritonic chemistry, ultrafast spectroscopy, and numerical methods related to quantum dynamics.
Read more on our group homepage.
Polaritonic Chemistry and Light-Matter Materials
Gaining detailed control over chemical reactions has always been a chemists dream. Quantum coherent control has been pursuing this dream by using specially tailored light fields to control chemical reactions on an atomistic level. With the advancement of cavity quantum electrodynamics and its recent application to molecules, using the quantum properties of light to control photo-chemistry has come into reach. Recent,
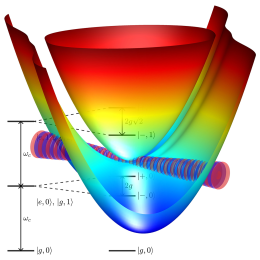
ground breaking experiments have show that one can utilize the vacuum field of an optical nano-resonator to significantly modify the potential energy landscape and thus its photo-chemistry. The underlying effect is the formation of so called "dressed states", which are created when the quantized radiation field mode couples to a molecular electronic transition. In the resulting coupled light-matter system the molecular and the photonic degrees of freedom are heavily mixed. While this effect is well understood for atomic samples, it is not yet fully understood for molecules. The introduction of the nuclear degrees of freedom requires new theoretical frameworks. This effect can be used to modify reaction pathways of chemical and photo-chemical reactions. This opens a wide range of possibilities to engineer novel types of light driven catalysts. We are looking at the underlying mechanisms and are working on building a suitable tool chest for numerical simulations. With the new insight and tools we want to propose new photo-chemical applications.
Ultrafast X-Ray Spectroscopy of Conical Intersections
Conical intersections (CoIns) so far have eluded direct experimental observations. The evidence for their existence is based on ultra fast relaxation rates and other indirect signatures. The rapidly varying energy gap in the vicinity of a CoIn poses a main obstacle for their direct detection. The required extreme combination of temporal and spectral resolution is not available in conventional optical femtosecond experiments. Ultra short laser pulses in the extreme ultraviolet and X-ray laser regime, as they are provided by free electron laser and high harmonic generation sources, fulfill the spectral and temporal requirements to resolve the coupled nuclear+electronic dynamics in the vicinity of CoIns. Ultrafast hard X-ray sources make time-resolved diffraction experiments possible, paving the way to capture the nuclear dynamics of molecules in time as well as in space, with the "molecular movie" of a CoIn as the ultimate goal.
We theoretically investigate novel, X-Ray based experimental techniques for spectroscopic detection of CoIns with ultra short X-ray pulses. Simulation strategies for non-linear X-ray spectra and diffraction schemes are applied to molecular system of increasing complexity.
Read more on our group homepage.
Forskningsprojekt
Publikationer
I urval från Stockholms universitets publikationsdatabas
-
Läs mer om Multidimensional high-harmonic echo spectroscopy: Resolving coherent electron dynamics in the EUV regime
Multidimensional high-harmonic echo spectroscopy: Resolving coherent electron dynamics in the EUV regime
2024. Shicheng Jiang (et al.). Proceedings of the National Academy of Sciences of the United States of America 121 (7)
Artikel -
Ab Initio Vibro-Polaritonic Spectra in Strongly Coupled Cavity-Molecule Systems
2023. Thomas Schnappinger, Markus Kowalewski. Journal of Chemical Theory and Computation 19 (24), 9278-9289
ArtikelLäs mer om Ab Initio Vibro-Polaritonic Spectra in Strongly Coupled Cavity-Molecule SystemsRecent experiments have revealed the profound effect of strong light–matter interactions in optical cavities on the electronic ground state of molecular systems. This phenomenon, known as vibrational strong coupling, can modify reaction rates and induce the formation of molecular vibrational polaritons, hybrid states involving both photon modes, and vibrational modes of molecules. We present an ab initio methodology based on the cavity Born–Oppenheimer Hartree–Fock ansatz, which is specifically powerful for ensembles of molecules, to calculate vibro-polaritonic IR spectra. This method allows for a comprehensive analysis of these hybrid states. Our semiclassical approach, validated against full quantum simulations, reproduces key features of the vibro-polaritonic spectra. The underlying analytic gradients also allow for optimization of cavity-coupled molecular systems and performing semiclassical dynamics simulations.
-
Cavity Born–Oppenheimer Hartree–Fock Ansatz: Light–Matter Properties of Strongly Coupled Molecular Ensembles
2023. Thomas Schnappinger (et al.). Journal of Physical Chemistry Letters 14 (36), 8024-8033
ArtikelLäs mer om Cavity Born–Oppenheimer Hartree–Fock AnsatzExperimental studies indicate that optical cavities can affect chemical reactions through either vibrational or electronic strong coupling and the quantized cavity modes. However, the current understanding of the interplay between molecules and confined light modes is incomplete. Accurate theoretical models that take into account intermolecular interactions to describe ensembles are therefore essential to understand the mechanisms governing polaritonic chemistry. We present an ab initio Hartree–Fock ansatz in the framework of the cavity Born–Oppenheimer approximation and study molecules strongly interacting with an optical cavity. This ansatz provides a nonperturbative, self-consistent description of strongly coupled molecular ensembles, taking into account the cavity-mediated dipole self-energy contributions. To demonstrate the capability of the cavity Born–Oppenheimer Hartree–Fock ansatz, we study the collective effects in ensembles of strongly coupled diatomic hydrogen fluoride molecules. Our results highlight the importance of the cavity-mediated intermolecular dipole–dipole interactions, which lead to energetic changes of individual molecules in the coupled ensemble.
-
Cavity-Modified Chemiluminescent Reaction of Dioxetane
2023. Mahesh Gudem, Markus Kowalewski. Journal of Physical Chemistry A 127 (45), 9483-9494
ArtikelLäs mer om Cavity-Modified Chemiluminescent Reaction of DioxetaneChemiluminescence is a thermally activated chemical process that emits a photon of light by forming a fraction of products in the electronic excited state. A well-known example of this spectacular phenomenon is the emission of light in the firefly beetle, where the formation of a four-membered cyclic peroxide compound and subsequent dissociation produce a light-emitting product. The smallest cyclic peroxide, dioxetane, also exhibits chemiluminescence but with a low quantum yield as compared to that of firefly dioxetane. Employing the strong light–matter coupling has recently been found to be an alternative strategy to modify the chemical reactivity. In the presence of an optical cavity, the molecular degrees of freedom greatly mix with the cavity mode to form hybrid cavity–matter states called polaritons. These newly generated hybrid light–matter states manipulate the potential energy surfaces and significantly change the reaction dynamics. Here, we theoretically investigate the effects of a strong light–matter interaction on the chemiluminescent reaction of dioxetane using the extended Jaynes–Cummings model. The cavity couplings corresponding to the electronic and vibrational degrees of freedom have been included in the interaction Hamiltonian. We explore how the cavity alters the ground- and excited-state path energy barriers and reaction rates. Our results demonstrate that the formation of excited-state products in the dioxetane decomposition process can be either accelerated or suppressed, depending on the molecular orientation with respect to the cavity polarization.
-
Machine-learned correction to ensemble-averaged wave packet dynamics
2023. Yannick Holtkamp (et al.). Journal of Chemical Physics 159 (9)
ArtikelLäs mer om Machine-learned correction to ensemble-averaged wave packet dynamicsFor a detailed understanding of many processes in nature involving, for example, energy or electron transfer, the theory of open quantumsystems is of key importance. For larger systems, an accurate description of the underlying quantum dynamics is still a formidable task, and,hence, approaches employing machine learning techniques have been developed to reduce the computational effort of accurate dissipativequantum dynamics. A downside of many previous machine learning methods is that they require expensive numerical training datasets forsystems of the same size as the ones they will be employed on, making them unfeasible to use for larger systems where those calculationsare still too expensive. In this work, we will introduce a new method that is implemented as a machine-learned correction term to the socalled Numerical Integration of Schrödinger Equation (NISE) approach. It is shown that this term can be trained on data from small systemswhere accurate quantum methods are still numerically feasible. Subsequently, the NISE scheme, together with the new machine-learnedcorrection, can be used to determine the dissipative quantum dynamics for larger systems. Furthermore, we show that the newly proposedmachine-learned correction outperforms a previously handcrafted one, which, however, improves the results already considerably.
-
The role of dephasing for dark state coupling in a molecular Tavis–Cummings model
2023. Eric Davidsson, Markus Kowalewski. Journal of Chemical Physics 159 (4)
ArtikelLäs mer om The role of dephasing for dark state coupling in a molecular Tavis–Cummings modelThe collective coupling of an ensemble of molecules to a light field is commonly described by the Tavis–Cummings model. This modelincludes numerous eigenstates that are optically decoupled from the optically bright polariton states. Accessing these dark states requiresbreaking the symmetry in the corresponding Hamiltonian. In this paper, we investigate the influence of non-unitary processes on the darkstate dynamics in the molecular Tavis–Cummings model. The system is modeled with a Lindblad equation that includes pure dephasing, as itwould be caused by weak interactions with an environment, and photon decay. Our simulations show that the rate of pure dephasing, as wellas the number of two-level systems, has a significant influence on the dark state population.
-
Nonadiabatic Wave Packet Dynamics with Ab Initio Cavity-Born-Oppenheimer Potential Energy Surfaces
2023. Thomas Schnappinger, Markus Kowalewski. Journal of Chemical Theory and Computation 19 (2), 460-471
ArtikelLäs mer om Nonadiabatic Wave Packet Dynamics with Ab Initio Cavity-Born-Oppenheimer Potential Energy SurfacesStrong coupling of molecules with quantized electromagnetic fields can reshape their potential energy surfaces by forming dressed states. In such a scenario, it is possible to manipulate the dynamics of the molecule and open new photochemical reaction pathways. A theoretical approach to describe such coupled molecular-photon systems is the Cavity-Born-Oppenheimer (CBO) approximation. Similarly to the standard Born-Oppenheimer (BO) approximation, the system is partitioned and the electronic part of the system is treated quantum mechanically. This separation leads to CBO surfaces that depend on both nuclear and photonic coordinates. In this work, we demonstrated, for two molecular examples, how the concept of the CBO approximation can be used to perform nonadiabatic wave packet dynamics of a coupled molecular-cavity system. The light-matter interaction is incorporated in the CBO surfaces and the associated nonadiabatic coupling elements. We show that molecular and cavity contributions can be treated on the same numerical footing. This approach gives a new perspective on the description of light-matter coupling in molecular systems.
-
Tracking Conical Intersections with Nonlinear X-ray Raman Spectroscopy
2022. Deependra Jadoun, Markus Kowalewski. Ultrafast Science 2022
ArtikelLäs mer om Tracking Conical Intersections with Nonlinear X-ray Raman SpectroscopyConical intersections are formed when 2 or more electronic states become degenerate and give rise to ultrafast nonadiabatic processes such as radiation-less decay channels and geometric phase effects. The branching of nuclear wave packets near a conical intersection creates a coherent superposition of electronic states, which carries information about the energy difference of the involved states. X-ray Raman techniques have been proposed to observe the coherent superposition of the electronic states and to monitor the evolving electronic state separation. However, these techniques rely on the coherence generated as the wave packet passes through the conical intersection, and the electronic energy gap before the wave packet passes through the conical intersection is not tracked. In this paper, we theoretically demonstrate how a nonlinear Raman detection scheme can be used to gain further insight into the nonadiabatic dynamics in the vicinity of the conical intersection. We employ a combination of a resonant visible/infrared pulse and an off-resonant x-ray Raman probe to map the electronic state separation around the conical intersection. We demonstrate that this technique can achieve high contrast and is able to selectively probe the narrow electronic state separation around the conical intersection.
-
Time-resolved X-ray and XUV based spectroscopic methods for nonadiabatic processes in photochemistry
2022. Thomas Schnappinger (et al.). Chemical Communications 58 (92), 12763-12781
ArtikelLäs mer om Time-resolved X-ray and XUV based spectroscopic methods for nonadiabatic processes in photochemistryThe photochemistry of numerous molecular systems is influenced by conical intersections (CIs). These omnipresent nonadiabatic phenomena provide ultra-fast radiationless relaxation channels by creating degeneracies between electronic states and decide over the final photoproducts. In their presence, the Born-Oppenheimer approximation breaks down, and the timescales of the electron and nuclear dynamics become comparable. Due to the ultra-fast dynamics and the complex interplay between nuclear and electronic degrees of freedom, the direct experimental observation of nonadiabatic processes close to CIs remains challenging. In this article, we give a theoretical perspective on novel spectroscopic techniques capable of observing clear signatures of CIs. We discuss methods that are based on ultra-short laser pulses in the extreme ultraviolet and X-ray regime, as their spectral and temporal resolution allow for resolving the ultra-fast dynamics near CIs.
-
Probing nonadiabatic dynamics with attosecond pulse trains and soft x-ray Raman spectroscopy
2022. Lorenzo Restaino, Deependra Jadoun, Markus Kowalewski. Structural Dynamics 9 (3)
ArtikelLäs mer om Probing nonadiabatic dynamics with attosecond pulse trains and soft x-ray Raman spectroscopyLinear off-resonant x-ray Raman techniques are capable of detecting the ultrafast electronic coherences generated when a photoexcited wave packet passes through a conical intersection. A hybrid femtosecond or attosecond probe pulse is employed to excite the system and stimulate the emission of the signal photon, where both fields are components of a hybrid pulse scheme. In this paper, we investigate how attosecond pulse trains, as provided by high-harmonic generation processes, perform as probe pulses in the framework of this spectroscopic technique, instead of single Gaussian pulses. We explore different combination schemes for the probe pulse as well as the impact of parameters of the pulse trains on the signals. Furthermore, we show how Raman selection rules and symmetry consideration affect the spectroscopic signal, and we discuss the importance of vibrational contributions to the overall signal. We use two different model systems, representing molecules of different symmetries, and quantum dynamics simulations to study the difference in the spectra. The results suggest that such pulse trains are well suited to capture the key features associated with the electronic coherence.
-
Triplet-triplet Annihilation Dynamics of Naphthalene
2022. Mahesh Gudem, Markus Kowalewski. Chemistry - A European Journal 28 (40)
ArtikelLäs mer om Triplet-triplet Annihilation Dynamics of NaphthaleneTriplet-triplet annihilation (TTA) is a spin-allowed conversion of two triplet states into one singlet excited state, which provides an efficient route to generate a photon of higher frequency than the incident light. Multiple energy transfer steps between absorbing (sensitizer) and emitting (annihilator) molecular species are involved in the TTA based photon upconversion process. TTA compounds have recently been studied for solar energy applications, even though the maximum upconversion efficiency of 50 % is yet to be achieved. With the aid of quantum calculations and based on a few key requirements, several design principles have been established to develop the well-functioning annihilators. However, a complete molecular level understanding of triplet fusion dynamics is still missing. In this work, we have employed multi-reference electronic structure methods along with quantum dynamics to obtain a detailed and fundamental understanding of TTA mechanism in naphthalene. Our results suggest that the TTA process in naphthalene is mediated by conical intersections. In addition, we have explored the triplet fusion dynamics under the influence of strong light-matter coupling and found an increase of the TTA based upconversion efficiency.
-
Suppressing non-radiative decay of photochromic organic molecular systems in the strong coupling regime
2022. Rafael C. Couto, Markus Kowalewski. Physical Chemistry, Chemical Physics - PCCP 24 (32), 19199-19208
ArtikelLäs mer om Suppressing non-radiative decay of photochromic organic molecular systems in the strong coupling regimeThe lifetimes of electronic excited states have a strong influence on the efficiency of organic solar cells. However, in some molecular systems a given excited state lifetime is reduced due to the non-radiative decay through conical intersections. Several strategies may be used to suppress this decay channel. The use of the strong light-matter coupling provided in optical nano-cavities is the focus of this paper. Here, we consider the meso–tert-butyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene molecule (meso–tert-butyl-BODIPY) as a showcase of how strong and ultrastrong coupling might help in the development of organic solar cells. The meso–tert-butyl-BODIPY is known for its low fluorescence yield caused by the non-radiative decay through a conical intersection. However, we show here that, by considering this system within a cavity, the strong coupling can lead to significant changes in the multidimensional landscape of the potential energy surfaces of meso–tert-butyl-BODIPY, suppressing almost completely the decay of the excited state wave packet back to the ground state. By means of multi configuration electronic structure calculations and nuclear wave packet dynamics, the coupling with the cavity is analyzed in-depth to provide further insight of the interaction. By fine-tuning the cavity field strength and resonance frequency, we show that one can change the nuclear dynamics in the excited state, and control the non-radiative decay. This may lead to a faster and more efficient population transfer or the suppression of it.
-
Photoinduced bond oscillations in ironpentacarbonyl give delayed synchronous bursts of carbonmonoxide release
2022. Ambar Banerjee (et al.). Nature Communications 13 (1)
ArtikelLäs mer om Photoinduced bond oscillations in ironpentacarbonyl give delayed synchronous bursts of carbonmonoxide releaseEarly excited state dynamics in the photodissociation of transition metal carbonyls determines the chemical nature of short-lived catalytically active reaction intermediates. However, time-resolved experiments have not yet revealed mechanistic details in the sub-picosecond regime. Hence, in this study the photoexcitation of ironpentacarbonyl Fe(CO)5 is simulated with semi-classical excited state molecular dynamics. We find that the bright metal-to-ligand charge-transfer (MLCT) transition induces synchronous Fe-C oscillations in the trigonal bipyramidal complex leading to periodically reoccurring release of predominantly axial CO. Metaphorically the photoactivated Fe(CO)5 acts as a CO geyser, as a result of dynamics in the potential energy landscape of the axial Fe-C distances and non-adiabatic transitions between manifolds of bound MLCT and dissociative metal-centered (MC) excited states. The predominant release of axial CO ligands and delayed release of equatorial CO ligands are explained in a unified mechanism based on the σ*(Fe-C) anti-bonding character of the receiving orbital in the dissociative MC states.
-
Sustainable packaging of quantum chemistry software with the Nix package manager
2022. Markus Kowalewski, Phillip Seeber. International Journal of Quantum Chemistry 122 (9)
ArtikelLäs mer om Sustainable packaging of quantum chemistry software with the Nix package managerThe installation of quantum chemistry software packages is commonly done manually and can be a time-consuming and complicated process. An update of the underlying Linux system requires a reinstallation in many cases and can quietly break software installed on the system. In this paper, we present an approach that allows for an easy installation of quantum chemistry software packages, which is also independent of operating system updates. The use of the Nix package manager allows building software in a reproducible manner, which allows for a reconstruction of the software for later reproduction of scientific results. The build recipes that are provided can be readily used by anyone to avoid complex installation procedures.
-
Time-Resolved Photoelectron Spectroscopy of Conical Intersections with Attosecond Pulse Trains
2021. Deependra Jadoun, Markus Kowalewski. Journal of Physical Chemistry Letters 12 (33), 8103-8108
ArtikelLäs mer om Time-Resolved Photoelectron Spectroscopy of Conical Intersections with Attosecond Pulse TrainsConical Intersections (CIs), which are believed to be ubiquitous in molecular and biological systems, open up ultrafast nonradiative decay channels. A superposition of electronic states is created when a molecule passes through a CI and the nuclear wave packet branches. The resulting electronic coherence can be considered a unique signature of the CI. The involved electronic states can be resolved in the energy domain with photoelectron spectroscopy using a femtosecond pulse as a probe. However, the observation of the created electronic coherence in the time domain requires probe pulses with several electron volts of bandwidth. Attosecond pulses can probe the electronic coherence but are unable to resolve the involved electronic states. In this Letter, we propose to address this restriction by using time- resolved photoelectron spectroscopy with an attosecond pulse train as a probe. We theoretically demonstrate that the resulting photoelectron spectrum may yield energy resolution as well as the information on the created coherences in the time domain.
-
Direct Transition from Triplet Excitons to Hybrid Light–Matter States via Triplet–Triplet Annihilation
2021. Markus Kowalewski (et al.). Journal of the American Chemical Society 143 (19), 7501-7508
ArtikelLäs mer om Direct Transition from Triplet Excitons to Hybrid Light–Matter States via Triplet–Triplet AnnihilationStrong light–matter coupling generates hybrid states that inherit properties of both light and matter, effectively allowing the modification of the molecular potential energy landscape. This phenomenon opens up a plethora of options for manipulating the properties of molecules, with a broad range of applications in photochemistry and photophysics. In this article, we use strong light–matter coupling to transform an endothermic triplet–triplet annihilation process into an exothermic one. The resulting gradual on–off photon upconversion experiment demonstrates a direct conversion between molecular states and hybrid light–matter states. Our study provides a direct evidence that energy can relax from nonresonant low energy molecular states directly into hybrid light–matter states and lays the groundwork for tunable photon upconversion systems that modify molecular properties in situ by optical cavities rather than with chemical modifications.
-
Capturing fingerprints of conical intersection
2021. Deependra Jadoun, Mahesh Gudem, Markus Kowalewski. Structural Dynamics 8
ArtikelLäs mer om Capturing fingerprints of conical intersectionMany recent experimental ultrafast spectroscopy studies have hinted at non-adiabatic dynamics indicating the existence of conical intersections, but their direct observation remains a challenge. The rapid change of the energy gap between the electronic states complicated their observation by requiring bandwidths of several electron volts. In this manuscript, we propose to use the combined information of different x-ray pump-probe techniques to identify the conical intersection. We theoretically study the conical intersection in pyrrole using transient x-ray absorption, time-resolved x-ray spontaneous emission, and linear off-resonant Raman spectroscopy to gather evidence of the curve crossing.
-
Controlling the Photostability of Pyrrole with Optical Nanocavities
2021. Mahesh Gudem, Markus Kowalewski. Journal of Physical Chemistry A 125 (5), 1142-1151
ArtikelLäs mer om Controlling the Photostability of Pyrrole with Optical NanocavitiesStrong light-matter coupling provides a new strategy to manipulate the non-adiabatic dynamics of molecules by modifying potential energy surfaces. The vacuum field of nanocavities can couple strongly with the molecular degrees of freedom and form hybrid light-matter states, termed as polaritons or dressed states. The photochemistry of molecules possessing intrinsic conical intersections can be significantly altered by introducing cavity couplings to create new conical intersections or avoided crossings. Here, we explore the effects of optical cavities on the photo-induced hydrogen elimination reaction of pyrrole. Wave packet dynamics simulations have been performed on the two-state, two-mode model of pyrrole, combined with the cavity photon mode. Our results show how the optical cavities assist in controlling the photostability of pyrrole and influence the reaction mechanism by providing alternative dissociation pathways. The cavity effects have been found to be intensely dependent on the resonance frequency. We further demonstrate the importance of the vibrational cavity couplings and dipole-self interaction terms in describing the cavity-modified non-adiabatic dynamics.
-
Multi-wave mixing in the high harmonic regime
2021. Shicheng Jiang, Markus Kowalewski, Konstantin E. Dorfman. Optics Express 29 (4), 4746-4754
ArtikelLäs mer om Multi-wave mixing in the high harmonic regimeIt has been demonstrated that electronic coherences across many eV can be detected in pump-probe experiments involving high harmonic sources. An additional degree of control over the phase matching can be employed by investigating a more general class of multi-wave mixing. Non-collinear multi-wave mixing of high harmonics with energy (q1ω1 + q2ω2) can be selectively detected along the direction of (q1k1 + q2k2). Simulations based on a recently developed semi-perturbative approach show that only the specific harmonic signals with q1ω1 close to the energy difference between ground state and excited states are observable when the two input pulses are well separated in time. The coherent dynamics between different states can be selectively tracked by detecting the time-delay dependent signals with different q1k1, which can overcome the potential spectral congestion in real experiments. Additionally, such non-collinear geometry can be used to separate the dephasing induced decay and collision induced recovery behaviors of pump-probe high harmonic signal typically observed in the time-resolved high harmonic pump-probe signals.
-
Simulating photodissociation reactions in bad cavities with the Lindblad equation
2020. Eric Davidsson, Markus Kowalewski. Journal of Chemical Physics 153 (23)
ArtikelLäs mer om Simulating photodissociation reactions in bad cavities with the Lindblad equationOptical cavities, e.g., as used in organic polariton experiments, often employ low finesse mirrors or plasmonic structures. The photon lifetime in these setups is comparable to the timescale of the nuclear dynamics governing the photochemistry. This highlights the need for including the effect of dissipation in the molecular simulations. In this study, we perform wave packet dynamics with the Lindblad master equation to study the effect of a finite photon lifetime on the dissociation of the MgH+ molecule model system. Photon lifetimes of several different orders of magnitude are considered to encompass an ample range of effects inherent to lossy cavities.
-
Atom Assisted Photochemistry in Optical Cavities
2020. Eric Davidsson, Markus Kowalewski. Journal of Physical Chemistry A 124 (23), 4672-4677
ArtikelLäs mer om Atom Assisted Photochemistry in Optical CavitiesStrong light-matter coupling can modify the photochemistry of molecular systems. The collective dynamics of an ensemble of molecules coupled to the light field plays a crucial role in experimental observations. However, the theory of polaritonic chemistry is primarily understood in terms of single molecules, since even in small molecular ensembles the collective dynamics becomes difficult to disentangle. Understanding of the underlying ensemble mechanisms is key to a conceptual understanding and interpretation of experiments. We present a model system that simplifies the problem by mixing two-level Mg atoms with a single MgH+ molecule and investigate its collective dynamics. Our focus is on the modified chemical properties of a single diatomic molecule in the presence of an ensemble of resonant atoms as well as the structure of the major and intermediate polariton states. We present quantum dynamics simulations of the coupled vibronic-photonic system for a variable size of the atomic ensemble. Special attention is given to dissociative the dynamics of the MgH+ molecule.
-
Ultrafast dynamics in the vicinity of quantum light-induced conical intersections
2019. András Csehi (et al.). New Journal of Physics 21
ArtikelLäs mer om Ultrafast dynamics in the vicinity of quantum light-induced conical intersectionsNonadiabatic effects appear due to avoided crossings or conical intersections (CIs) that are either intrinsic properties in field-free space or induced by a classical laser field in a molecule. It was demonstrated that avoided crossings in diatomics can also be created in an optical cavity. Here, the quantized radiation field mixes the nuclear and electronic degrees of freedom creating hybrid field-matter states called polaritons. In the present theoretical study we go further and create CIs in diatomics by means of a radiation field in the framework of cavity quantum electrodynamics. By treating all degrees of freedom, that is the rotational, vibrational, electronic and photonic degrees of freedom on an equal footing we can control the nonadiabatic quantum light-induced dynamics by means of CIs. First, the pronounced difference between the the quantum light-induced avoided crossing and the CI with respect to the nonadiabatic dynamics of the molecule is demonstrated. Second, we discuss the similarities and differences between the classical and the quantum field description of the light for the studied scenario.
-
Quantum control with quantum light of molecular nonadiabaticity
2019. András Csehi (et al.). Physical Review A. Atomic, Molecular, and Optical Physics 100 (5)
ArtikelLäs mer om Quantum control with quantum light of molecular nonadiabaticityCoherent control experiments in molecules are often done with shaped laser fields. The electric field is described classically and control over the time evolution of the system is achieved by shaping the laser pulses in the time or frequency domain. Moving on from a classical to a quantum description of the light field allows one to engineer the quantum state of light to steer chemical processes. The quantum field description of the photon mode allows one to manipulate the light-matter interaction directly in phase space. In this paper we demonstrate the basic principle of coherent control with quantum light on the avoided crossing in lithium fluoride. Using a quantum description of light together with the nonadiabatic couplings and vibronic degrees of freedoms opens up alternative perspective on quantum control. We show the deviations from control with purely classical light field and how back-action of the light field becomes important in a few-photon regime.
-
Monitoring nonadiabatic dynamics in molecules by ultrafast X-Ray diffraction
2019. Markus Kowalewski, Kochise Bennett, Shaul Mukamel. EPJ Web of Conferences 205
ArtikelLäs mer om Monitoring nonadiabatic dynamics in molecules by ultrafast X-Ray diffractionWe theoretically examine time-resolved diffraction from molecules which undergo non-adiabatic dynamics and identify contributions from inelastic scattering that indicate the presence of an avoided crossing and the corresponding nuclear configuration.
-
Imaging of transition charge densities involving carbon core excitations by all X-ray sum-frequency generation
2019. Daeheum Cho (et al.). Philosophical Transactions. Series A 377 (2145)
ArtikelLäs mer om Imaging of transition charge densities involving carbon core excitations by all X-ray sum-frequency generationX-ray diffraction signals from the time-evolving molecular charge density induced by selective core excitation of chemically inequivalent carbon atoms are calculated. A narrowband X-ray pulse selectively excites the carbon K-edge of the –CH3 or –CH2F groups in fluoroethane (CH3–CH2F). Each excitation creates a distinct core coherence which depends on the character of the electronic transition. Direct propagation of the reduced single-electron density matrix, using real-time time-dependent density functional theory, provides the time-evolving charge density following interactions with external fields. The interplay between partially filled valence molecular orbitals upon core excitation induces characteristic femtosecond charge migration which depends on the core–valence coherence, and is monitored by the sum-frequency generation diffraction signal.
Visa alla publikationer av Markus Kowalewski vid Stockholms universitet