Thomas Schnappinger
About me
Trained as a theoretical chemist, I work at the interface of physics and chemistry, investigating how light-matter interactions can influence and control (photo)chemical reactions.
Research
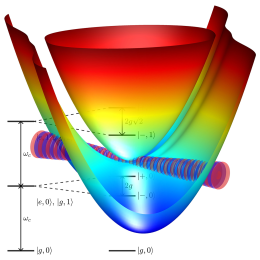
The interaction between light and matter is fundamental to how we perceive and understand the world. This interaction is omnipresent in our daily lives and is typically weak. In this context, "weak" means that light only perturbs matter without changing its properties. But when light is confined to small volumes, such as between two mirrors separated at the nanoscale, it reveals its quantum properties and fascinating quantum phenomena come into play. Under these conditions, photons can strongly interact with molecules, giving rise to hybrid light-matter states known as polaritons. Polaritons have attracted considerable attention because experiments have shown that they can modify molecular properties. Polaritonic chemistry is therefore a promising tool to fulfill the long-standing dream in chemistry to control reactions with light.
However, current theoretical models are able to capture only certain aspects and cannot fully explain experimental results. The main goal of my reaserch is to develop computational tools that can simulate polaritonic chemistry in realistic scenarios. The overall goal is to derive concepts for polaritonic chemistry based on chemical and physical intuition. Such rules are crucial for predicting which reactions can be modified by strong coupling, making experimental progress less dependent on trial and error.
Research projects
Publications
A selection from Stockholm University publication database
-
Do Molecular Geometries Change Under Vibrational Strong Coupling?
2024. Thomas Schnappinger, Markus Kowalewski. The Journal of Physical Chemistry Letters 15 (30), 7700-7707
ArticleRead more about Do Molecular Geometries Change Under Vibrational Strong Coupling?As pioneering experiments have shown, strong coupling between molecular vibrations and light modes in an optical cavity can significantly alter molecular properties and even affect chemical reactivity. However, the current theoretical description is limited and far from complete. To explore the origin of this exciting observation, we investigate how the molecular structure changes under strong light–matter coupling using an ab initio method based on the cavity Born–Oppenheimer Hartree–Fock ansatz. By optimizing H2O and H2O2 resonantly coupled to cavity modes, we study the importance of reorientation and geometric relaxation. In addition, we show that the inclusion of one or two cavity modes can change the observed results. On the basis of our findings, we derive a simple concept to estimate the effect of the cavity interaction on the molecular geometry using the molecular polarizability and the dipole moments.
-
Unraveling a Cavity-Induced Molecular Polarization Mechanism from Collective Vibrational Strong Coupling
2024. Dominik Sidler (et al.). The Journal of Physical Chemistry Letters 15 (19), 5208-5214
ArticleRead more about Unraveling a Cavity-Induced Molecular Polarization Mechanism from Collective Vibrational Strong CouplingWe demonstrate that collective vibrational strong coupling of molecules in thermal equilibrium can give rise to significant local electronic polarizations in the thermodynamic limit. We do so by first showing that the full nonrelativistic Pauli–Fierz problem of an ensemble of strongly coupled molecules in the dilute-gas limit reduces in the cavity Born–Oppenheimer approximation to a cavity–Hartree equation for the electronic structure. Consequently, each individual molecule experiences a self-consistent coupling to the dipoles of all other molecules, which amount to non-negligible values in the thermodynamic limit (large ensembles). Thus, collective vibrational strong coupling can alter individual molecules strongly for localized ”hotspots” within the ensemble. Moreover, the discovered cavity-induced polarization pattern possesses a zero net polarization, which resembles a continuous form of a spin glass (or better polarization glass). Our findings suggest that the thorough understanding of polaritonic chemistry, requires a self-consistent treatment of dressed electronic structure, which can give rise to numerous, so far overlooked, physical mechanisms.
-
Ab Initio Vibro-Polaritonic Spectra in Strongly Coupled Cavity-Molecule Systems
2023. Thomas Schnappinger, Markus Kowalewski. Journal of Chemical Theory and Computation 19 (24), 9278-9289
ArticleRead more about Ab Initio Vibro-Polaritonic Spectra in Strongly Coupled Cavity-Molecule SystemsRecent experiments have revealed the profound effect of strong light–matter interactions in optical cavities on the electronic ground state of molecular systems. This phenomenon, known as vibrational strong coupling, can modify reaction rates and induce the formation of molecular vibrational polaritons, hybrid states involving both photon modes, and vibrational modes of molecules. We present an ab initio methodology based on the cavity Born–Oppenheimer Hartree–Fock ansatz, which is specifically powerful for ensembles of molecules, to calculate vibro-polaritonic IR spectra. This method allows for a comprehensive analysis of these hybrid states. Our semiclassical approach, validated against full quantum simulations, reproduces key features of the vibro-polaritonic spectra. The underlying analytic gradients also allow for optimization of cavity-coupled molecular systems and performing semiclassical dynamics simulations.
-
Cavity Born–Oppenheimer Hartree–Fock Ansatz: Light–Matter Properties of Strongly Coupled Molecular Ensembles
2023. Thomas Schnappinger (et al.). The Journal of Physical Chemistry Letters 14 (36), 8024-8033
ArticleRead more about Cavity Born–Oppenheimer Hartree–Fock AnsatzExperimental studies indicate that optical cavities can affect chemical reactions through either vibrational or electronic strong coupling and the quantized cavity modes. However, the current understanding of the interplay between molecules and confined light modes is incomplete. Accurate theoretical models that take into account intermolecular interactions to describe ensembles are therefore essential to understand the mechanisms governing polaritonic chemistry. We present an ab initio Hartree–Fock ansatz in the framework of the cavity Born–Oppenheimer approximation and study molecules strongly interacting with an optical cavity. This ansatz provides a nonperturbative, self-consistent description of strongly coupled molecular ensembles, taking into account the cavity-mediated dipole self-energy contributions. To demonstrate the capability of the cavity Born–Oppenheimer Hartree–Fock ansatz, we study the collective effects in ensembles of strongly coupled diatomic hydrogen fluoride molecules. Our results highlight the importance of the cavity-mediated intermolecular dipole–dipole interactions, which lead to energetic changes of individual molecules in the coupled ensemble.
-
Nonadiabatic Wave Packet Dynamics with Ab Initio Cavity-Born-Oppenheimer Potential Energy Surfaces
2023. Thomas Schnappinger, Markus Kowalewski. Journal of Chemical Theory and Computation 19 (2), 460-471
ArticleRead more about Nonadiabatic Wave Packet Dynamics with Ab Initio Cavity-Born-Oppenheimer Potential Energy SurfacesStrong coupling of molecules with quantized electromagnetic fields can reshape their potential energy surfaces by forming dressed states. In such a scenario, it is possible to manipulate the dynamics of the molecule and open new photochemical reaction pathways. A theoretical approach to describe such coupled molecular-photon systems is the Cavity-Born-Oppenheimer (CBO) approximation. Similarly to the standard Born-Oppenheimer (BO) approximation, the system is partitioned and the electronic part of the system is treated quantum mechanically. This separation leads to CBO surfaces that depend on both nuclear and photonic coordinates. In this work, we demonstrated, for two molecular examples, how the concept of the CBO approximation can be used to perform nonadiabatic wave packet dynamics of a coupled molecular-cavity system. The light-matter interaction is incorporated in the CBO surfaces and the associated nonadiabatic coupling elements. We show that molecular and cavity contributions can be treated on the same numerical footing. This approach gives a new perspective on the description of light-matter coupling in molecular systems.
-
Time-resolved X-ray and XUV based spectroscopic methods for nonadiabatic processes in photochemistry
2022. Thomas Schnappinger (et al.). Chemical Communications 58 (92), 12763-12781
ArticleRead more about Time-resolved X-ray and XUV based spectroscopic methods for nonadiabatic processes in photochemistryThe photochemistry of numerous molecular systems is influenced by conical intersections (CIs). These omnipresent nonadiabatic phenomena provide ultra-fast radiationless relaxation channels by creating degeneracies between electronic states and decide over the final photoproducts. In their presence, the Born-Oppenheimer approximation breaks down, and the timescales of the electron and nuclear dynamics become comparable. Due to the ultra-fast dynamics and the complex interplay between nuclear and electronic degrees of freedom, the direct experimental observation of nonadiabatic processes close to CIs remains challenging. In this article, we give a theoretical perspective on novel spectroscopic techniques capable of observing clear signatures of CIs. We discuss methods that are based on ultra-short laser pulses in the extreme ultraviolet and X-ray regime, as their spectral and temporal resolution allow for resolving the ultra-fast dynamics near CIs.
Show all publications by Thomas Schnappinger at Stockholm University
$presentationText