Michael OdeliusProfessor
Om mig
Med en bakgrund i fysikalisk kemi, arbetar jag idag med forskning, undervisning och samverkan vid Fysikum. Jag studerar kemiska processer och molekyler med datorsimuleringar baserade på kvantkemiska metoder.
Utlysning av doktorandtjänst
Vi söker för närvarande kandidater för en doktorand i teoretisk kemisk fysik för undersökningar av energirika azidföreningar. Fotokemin of azider (R-N=N=N) ger upphov till en mängd olika möjligheter i organisk syntes och i området klick-kemi, som belönades med 2022 års Nobelpris i kemi.
Undervisning
Vid Fysikum på Stockholms universitet har jag de senaste åren haft föreläsningar på en kurs i kvantkemi (FK7059), som vi nyligen omvandlat till en kurs med förinspelade videoföreläsningar och mer aktivt lärande.
Om du är intresserad av ett examensarbete på kanditat- eller masterniv kan du hitta exempel på tidigare arbeten på hemsidan för avdelningen kemisk fysik. Välkommen att kontakta mig om du är nyfiken.
En viktig del i vårt arbete vid sidan av forskning och undervisning är att inspirera unga och stödja deras intresse för fysik, därför har vi skapat olika kontaktytor för skolor och allmänheten som våra öppna föreläsningar, evenemanget Fysik i Kungsträdgården och de senaste åren, Forskarfredag.
Forskning
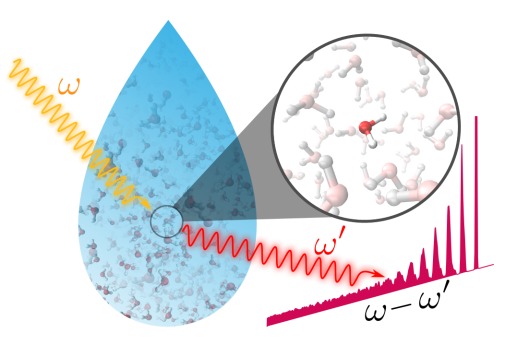

Vi använder teoretiska beräkningar baserade på kvantmekanik och statistisk fysik för att studera vätskor och solceller. Med dessa metoder studerar vi elektronstruktur och molekyldynamik på molekylär nivå. Genom datorsimuleringar kan vi i nära samarbete med experiment ge en detaljförståelse av komplicerade system, och exempelvis bidra till utvecklingen av effektivare kemisk lagring av solenergi.
På forskningsgruppens hemsida kan du läsa mer om pågående forskningsprojekt, och hitta exempel på tidigare publikationer.
Utlysning av doktorandtjänst
Vi söker för närvarande kandidater för en doktorand i teoretisk kemisk fysik för undersökningar av energirika azidföreningar. Fotokemin of azider (R-N=N=N) ger upphov till en mängd olika möjligheter i organisk syntes och i området klick-kemi, som belönades med 2022 års Nobelpris i kemi.
Forskningsprojekt


Publikationer
I urval från Stockholms universitets publikationsdatabas
-
Electronic Structure Changes of an Aromatic Amine Photoacid along the Förster Cycle
2022. Sebastian Eckert (et al.). Angewandte Chemie International Edition 61 (27)
ArtikelLäs mer om Electronic Structure Changes of an Aromatic Amine Photoacid along the Förster CyclePhotoacids show a strong increase in acidity in the first electronic excited state, enabling real-time studies of proton transfer in acid-base reactions, proton transport in energy storage devices and biomolecular sensor protein systems. Several explanations have been proposed for what determines photoacidity, ranging from variations in solvation free energy to changes in electronic structure occurring along the four stages of the Forster cycle. Here we use picosecond nitrogen K-edge spectroscopy to monitor the electronic structure changes of the proton donating group in a protonated aromatic amine photoacid in solution upon photoexcitation and subsequent proton transfer dynamics. Probing core-to-valence transitions locally at the amine functional group and with orbital specificity, we clearly reveal pronounced electronic structure, dipole moment and energetic changes on the conjugate photobase side. This result paves the way for a detailed electronic structural characterization of the photoacidity phenomenon.
-
A-site cation influence on the conduction band of lead bromide perovskites
2022. Gabriel J. Man (et al.). Nature Communications 13 (1)
ArtikelLäs mer om A-site cation influence on the conduction band of lead bromide perovskitesHot carrier solar cells hold promise for exceeding the Shockley-Queisser limit. Slow hot carrier cooling is one of the most intriguing properties of lead halide perovskites and distinguishes this class of materials from competing materials used in solar cells. Here we use the element selectivity of high-resolution X-ray spectroscopy and density functional theory to uncover a previously hidden feature in the conduction band states, the sigma-pi energy splitting, and find that it is strongly influenced by the strength of electronic coupling between the A-cation and bromide-lead sublattice. Our finding provides an alternative mechanism to the commonly discussed polaronic screening and hot phonon bottleneck carrier cooling mechanisms. Our work emphasizes the optoelectronic role of the A-cation, provides a comprehensive view of A-cation effects in the crystal and electronic structures, and outlines a broadly applicable spectroscopic approach for assessing the impact of chemical alterations of the A-cation on perovskite electronic structure. The A-cation influence on the mechanism of slow hot carrier cooling in perovskites is controversial. Here, Man et al. resolve a debated issue regarding A-cation influence on the electronic structure of lead halide perovskites.
-
Spatial microheterogeneity in the valence band of mixed halide hybrid perovskite materials
2022. Axel Erbing (et al.). Chemical Science 13 (32), 9285-9294
ArtikelLäs mer om Spatial microheterogeneity in the valence band of mixed halide hybrid perovskite materialsThe valence band of lead halide hybrid perovskites with a mixed I/Br composition is investigated using electronic structure calculations and complementarily probed with hard X-ray photoelectron spectroscopy. In the latter, we used high photon energies giving element sensitivity to the heavy lead and halide ions and we observe distinct trends in the valence band as a function of the I : Br ratio. Through electronic structure calculations, we show that the spectral trends with overall composition can be understood in terms of variations in the local environment of neighboring halide ions. From the computational model supported by the experimental evidence, a picture of the microheterogeneity in the valence band maximum emerges. The microheterogeneity in the valence band suggests that additional charge transport mechanisms might be active in lead mixed halide hybrid perovskites, which could be described in terms of percolation pathways.
-
Photoinduced bond oscillations in ironpentacarbonyl give delayed synchronous bursts of carbonmonoxide release
2022. Ambar Banerjee (et al.). Nature Communications 13 (1)
ArtikelLäs mer om Photoinduced bond oscillations in ironpentacarbonyl give delayed synchronous bursts of carbonmonoxide releaseEarly excited state dynamics in the photodissociation of transition metal carbonyls determines the chemical nature of short-lived catalytically active reaction intermediates. However, time-resolved experiments have not yet revealed mechanistic details in the sub-picosecond regime. Hence, in this study the photoexcitation of ironpentacarbonyl Fe(CO)5 is simulated with semi-classical excited state molecular dynamics. We find that the bright metal-to-ligand charge-transfer (MLCT) transition induces synchronous Fe-C oscillations in the trigonal bipyramidal complex leading to periodically reoccurring release of predominantly axial CO. Metaphorically the photoactivated Fe(CO)5 acts as a CO geyser, as a result of dynamics in the potential energy landscape of the axial Fe-C distances and non-adiabatic transitions between manifolds of bound MLCT and dissociative metal-centered (MC) excited states. The predominant release of axial CO ligands and delayed release of equatorial CO ligands are explained in a unified mechanism based on the σ*(Fe-C) anti-bonding character of the receiving orbital in the dissociative MC states.
-
Core-Level Binding Energy Reveals Hydrogen Bonding Configurations of Water Adsorbed on TiO2 (110) Surface
2021. Chinnathambi Kamal (et al.). Physical Review Letters 126 (1)
ArtikelLäs mer om Core-Level Binding Energy Reveals Hydrogen Bonding Configurations of Water Adsorbed on TiO2 (110) SurfaceUsing x-ray photoelectron spectroscopy of the oxygen 1s core level, the ratio between intact (D2O) and dissociated (OD) water in the hydrated stoichiometric TiO2 (110) surface is determined at varying coverage and temperature. In the submonolayer regime, both the D2O:OD ratio and the core-level binding energy of D2O (Delta BE) decrease with temperature. The observed variations in Delta BE are shown with density functional theory to be governed crucially and solely by the local hydrogen bonding environment, revealing a generally applicable classification and details about adsorption motifs.
Visa alla publikationer av Michael Odelius vid Stockholms universitet